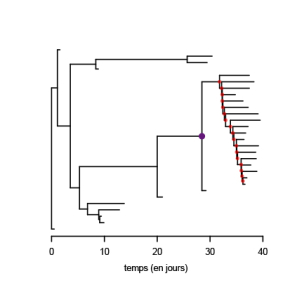
La super-propagation dans les génomes
On s’interroge beaucoup sur l’utilité du séquençage des virus SARS-CoV-2 de patients infectés. Une perspective rédigée par un chercheur du laboratoire Maladies Infectieuses et Vecteurs: Écologie, Génétique, Évolution et Contrôle (MIVEGEC – CNRS / IRD / Université de Montpellier) et parue dans la revue Science décortique comment l’analyse de ces génomes permet de détecter et comprendre des événements de « super-propagation » dans lesquels une personne en infecte de nombreuses autres. L’analyse de séquences est potentiellement moins chère que le suivi de contact, mais elle est aussi moins intrusive.
On pense que les événements de super-propagation pourraient être un des facteurs essentiels à la propagation du SARS-CoV-2. Si de tels événements sautent aux yeux quand on analyse des données de suivi de contact, déterminer leur fréquence et leur effet quantitatif sur l’épidémie de COVID-19 est bien plus délicat. De plus, les données de suivi de contact sont chères à collecter, biaisées et souvent de faible qualité (les personnes répondant n’étant pas sûres de l’origine de leur contamination). Le traçage numérique pourrait répondre à ces biais mais soulève des risques énormes en termes de libertés publiques. Les données issues du séquençage des génomes de virus infectant des patients offrent une alternative intéressante qui minimise à la fois les biais d’analyse et les risques éthiques.
Comme tous les virus, le SARS-CoV-2 évolue et, ce faisant, ses populations accumulent des mutations (environ 1 à 2 par mois par génome en moyenne). Une des conséquences est que des personnes proches dans la chaîne de transmission sont a priori infectées par des virus dont les génomes se ressemblent. Des chercheurs de Boston ont ainsi réussi à montrer, grâce à un échantillonnage à la fois dense et précoce dans l’épidémie, qu’un centre de conférences d’affaires a été la source d’un événement de super-propagation qui a eu des ramifications au niveau national et même international. En effet, la base de données GISAID leur a permis de conclure que cet événement était potentiellement associé à plus de 100.000 contaminations de par le monde. De plus, ils ont pu détecter un second événement de propagation dans un centre de soins. Ce dernier était intéressant à double titre. D’une part, ils ont pu identifier deux introductions du virus dans le centre de soin, mais seule une des deux a conduit à de la super-propagation. D’autre part, contrairement au centre de conférences d’affaire, l’événement de super-propagation dans le centre de soin a eu peu de ramification dans le reste de la ville, mais a été associé à une mortalité élevée dans le centre.
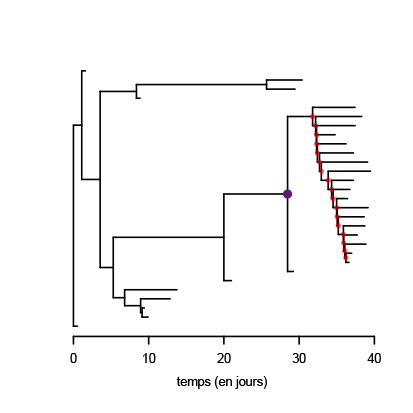
La génomique représente donc un outil sous-exploité pour mieux comprendre la nature des événements de propagation, et, par exemple, si ceux-ci sont liés au contexte environnemental, au comportement individuel (nombre de contacts) ou à la biologie de l’infection (charge virale). De nombreux défis demeurent pour intégrer ces données de séquences aux données classiques d’incidence (nombre de nouveaux cas) utilisées en routine. Mais une telle combinaison de données pourrait se révéler particulièrement précieuse dans le cas du SARS-CoV-2 qui accumule moins de mutations dans son génome que des virus tels que le VIH.
Objectifs de développement durable

- ODD 3 : bonne santé et bien-être
Référence
Alizon S. (2021) Superspreading genomes. Science. 371(6529):574–575, doi: 10.1126/science.abg0100